47 хромосом и маркерная
Содержание
Лишние хромосомы у мужчин, 47 хромосом, синдром XYY
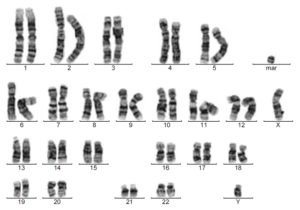
Лишняя хромосома у мальчиков, синдром XYY – это группа симптомов, которая поражает мужчин. У некоторых признаки едва заметны. Общие проявления включают в себя трудности обучения, задержку речи, низкий мышечный тонус (гипотония).
Вызван дополнительной копией Y хромосомы в каждой клетке тела, возникает случайно, не наследуется. Диагноз может быть сделан на основании пренатальных тестов, или в детстве, зрелом возрасте, если у мужчины есть симптомы заболевания.
Лечение включает специальное образование, терапию при задержках развития.
Другие имена:
- 47, синдром XYY;
- Синдром Иакова;
- XYY-кариотип;
- Синдром YY.
Характеристики
Хyy синдром- редкое хромосомное расстройство. Мужчины обычно имеют одну Х и Y-хромосому. Однако у людей с этим синдромом есть одна Х и две Y. Затронутые люди обычно очень высокие. В подростковом возрасте многие имеют значительную угревую сыпь.
Дополнительные симптомы:
- проблемы с обучением;
- поведенческие трудности, такие как импульсивность.
Интеллект обычно находится в нормальном диапазоне, хотя IQ в среднем на 10-15 баллов ниже, чем у здоровых людей.
В прошлом было много неправильных представлений об этой болезни. Ее иногда называли супер-мужским заболеванием, потому что люди с этим расстройством считались чрезмерно агрессивными, не имели сочувствия. Недавние исследования показали, что это не так.
Первые годы обучения в школе могут быть более сложными для мальчиков с синдромом XYY, но они обычно продолжают вести полноценную, здоровую, нормальную жизнь.
Симптомы
Симптомы при наличии лишней хромосомы могут варьироваться от едва заметных до более серьезных. Считается, что некоторые мужчины с никогда не бывают диагностированы, потому что признаки не заметны.
Для других обычен: низкий мышечный тонус (гипотония), задержка речи (в позднем младенчестве, раннем детстве). Некоторые мальчики с синдромом XYY испытывают трудности по некоторым предметам в школе, таким как чтение, письмо.
Обычно нет интеллектуальной недееспособности, хотя среднее IQ на 10-15 баллов ниже чем у обычного человека.
Другие признаки: астма; проблемы с зубами; тяжелые кистозные угри в подростковом возрасте. У мальчиков с расстройством обычно нет физических особенностей, отличных от большинства людей, но они могут быть выше, чем ожидалось.
Некоторые мужчины с лишней хромосомой имеют поведенческие различия, такие как расстройство спектра аутизма (обычно на более мягком уровне) или синдром дефицита внимания с гиперактивностью (ADHD).
Существует повышенный риск возникновения тревожных расстройств или расстройств настроения.
Большинство проходят нормальное половое развитие.
Однако у некоторых может развиться протекание яичка (когда яички не производят сперму или тестостерон), что приводит к проблемам бесплодия.
В некоторых случаях у затронутых людей возникают такие поведенческие проблемы, как взрывной характер, гиперактивность, импульсивность, вызывающие действия, антиобщественное поведение.Ниже перечислены симптомы, которые могут иметь люди с этой болезнью.
| Медицинские термины | Другие имена |
| Отсроченная речь, ЗРР | |
| Низкопосаженные уши | |
| Задержка моторного развития | |
| Высокий рост | |
| Удушье | |
| Синдром дефицита внимания и гиперактивности | Недостатки внимания |
| Врожденная ночная слепота | |
| Пальцы клинодактильно | |
| Гипертелоризм | Широко расставленные глаза |
| Нарушение социальных взаимодействий | Плохие социальные навыки |
| Импульсивность | |
| Интеллектуальная недееспособность | Умственная отсталость |
| Макроцефалия | Увеличенный размер черепа |
| Неонатальная гипотония | Низкий мышечный тонус в неонатальном периоде |
| Конкретная обучающая инвалидность | |
| Аномальная форма ствола мозга | |
| Азооспермия | Отсутствие спермы |
| Дисфункции мозжечка | |
| Крипторхизм | Неопущенные семенники |
| Дисгенезис мозжечка | |
| Гидроцефалия | Много спинномозговой жидкости в мозге |
| Гипоспадии | |
| Увеличение уровня гонадотропина | |
| Повышенный уровень тестостерона | |
| Macroorchidism | Большие яички |
| Мужское бесплодие | |
| Микропенис | Короткий пенис |
| Oligospermia | Низкое количество сперматозоидов |
| Судороги | Припадки |
| Варикоцеле |
Узнать больше Мукополисахаридоз iii типа при синдроме санфилиппо
Причина
XYY синдром вызван наличием дополнительной копии Y хромосома в каждой клетке тела. У большинства людей две половые хромосомы, у девочек две Х, мальчиков – одна Х и одна Y.
Обычно мужчины имеют 46 хромосом, включая одну Х и одну Y. Мужчины с лишней хромосомой имеют 47, две из которых являются Y. Большинство случаев синдрома XYY обусловлены ошибкой деления клеток в сперме до зачатия. Редко, ошибка деления ячейки возникает после зачатия, приводит к мозаике клеток с 46 хромосомами и 47 хромосомами.
Точная причина, по которой происходят эти мутации в делении клеток не поняты.
Не совсем понятно, почему дополнительная копия Y-хромосомы вызывает функции, связанные с синдромом хуу. Считается, что высокий рост, наблюдаемый у некоторых мужчин с расстройством, вызван наличием дополнительной копии гена SHOX.
Этот ген дает инструкции организму контролировать рост костей.
Люди, у которых есть дополнительная Y-хромосома, имеют дополнительную копию гена SHOX, что объясняет, почему они выше, чем ожидалось.
Другой ген, который, вызывает симптомы называется NLGN4Y. Он расположен на Y-хромосоме, предоставляет инструкции для тела, помогает формировать связи между клетками головного мозга. Считается, что наличие такой дополнительной копии вызывает проблемы с обучением
Затронутые популяции
Это редкое хромосомное расстройство, присутствующее при рождении, которое поражает только мужчин. Происходит примерно у одного из 1000.
Наследование
XYY синдром обычно не унаследован от родителя. Вызван случайным событием, которое происходит во время формирования сперматозоидов до зачатия.
Если у пары есть ребенок с хуу синдромом, шансы на то, они будут иметь другого ребенка с мутацией, не увеличиваются. Мужчины с лишней хромосомой не имеют повышенный риск передать патологию потомству.
Все вопросы о возможности иметь ребенка с хромосомной аномалией необходимо задать специалисту по генетике.
Диагностика
Диагностика производится на основе тщательной клинической оценки, детальной истории пациентов и специализированных тестов (Хромосомного анализа), которые обнаруживают наличие дополнительной Y-хромосомы (47, XYY-кариотип).
hy диагноз подозревается, когда врач наблюдает признаки, связанные с синдромом, такие как низкий мышечный тонус (гипотония), задержки речи, проблемы обучения в школе. Затем проводится тестирование, чтобы узнать, есть ли генетическое объяснение симптомов.
Рекомендуемые исследования:
- Кариотип: тест, который используется для просмотра всех хромосом в клетке;
- Хромосомный микрочип: ищет дополнительные или отсутствующие хромосомы, или их фрагменты.
В некоторых случаях синдром 47 хромосомы может подозреваться пренатально на основе обычного скрининга. Диагноз подтверждается пренатальными тестами, такими как амниоцентез или выбор хорионических ворсинок (CVS).
Оценка речи и языка должна происходить в течение первых 24 месяцев. исследование чтения проводят в школьном возрасте, чтобы исключить дислексию. Поведенческое тестирование применимо при импульсивности, плохом внимании, проблемах в социальных навыках.
Считается, что некоторые мужчины с лишней хромосомой никогда не диагностируются, потому что нет серьезных проявлений болезни.
Узнать больше Что такое синдром Драве?
Связанные нарушения
Симптомы следующих заболеваний могут быть сходными:
Синдром Клайнфелтера
Относится к группе хромосомных расстройств у мужчин, при которых присутствует одна или несколько дополнительных Х-хромосом. Мужчины с классической формой расстройства имеют одну дополнительную Х-хромосому.
Люди с вариантными формами синдрома Клайнфелтера имеют дополнительные Х или Y-хромосомы. Дополнительная хромосома влияет на физическое, развивающее, поведенческое и когнитивное функционирование. Общие физические особенности:
- высокий рост;
- отсутствие вторичного развития пубертата;
- небольшие яички (гипогонадизм);
- задержка развития пубертата и развитие груди (гинекомастия) в позднем периоде полового созревания.
Эти особенности связаны с низким уровнем тестостерона и повышенными уровнями гонадотропина.
Синдром Сотоса
Переменное генетическое заболевание, характеризующееся чрезмерным ростом до и после рождения. Одной из основных особенностей синдрома Сотоса является особая внешность лица, которая включает аномально выраженный лоб (лобное боссу), наклонные складчатые веки (пальпебральные трещины), видную узкую челюсть, длинное узкое лицо, форму головы похожую на перевернутую грушу.
Высота и окружность головы больше, чем средняя для большинства пострадавших. Задержка развития присутствует у большинства детей с синдромом Сотоса. Она включает моторные и языковые задержки, умственную отсталость.
Другие проблемы: желтуха у новорожденных, изогнутый позвоночник (сколиоз), судороги, косоглазие, потеря слуха, врожденные пороки сердца, аномалии почек, поведенческие проблемы. У затронутых лиц повышен риск развития определенных типов опухолей.
Расстройство происходит из-за аномалий гена NSD1.
Синдром Марфана
Генетическое заболевание, поражающее соединительную ткань, которая придает тканям форму и силу. Соединительная ткань находится по всему телу. У многих пациентов с синдромом Марфана поражаются многочисленные системы органов. Сердечные и кровеносные сосуды, опорно-двигательная система, глаза поражаются чаще всего. Основные симптомы:
- чрезмерный рост костей рук, ног;
- аномальная боковая кривизна позвоночника (сколиоз);
- углубление или выпячивание грудной стенки (pectus);
- дислокация глаз (эктопия lentis);
- близорукость;
- расширение (аневризма) и разрыв (рассечение) аорты;
- гибкость митрального клапана (пролапс митрального клапана);
- обратный поток крови через аортальный и митральный клапаны (аортальная, митральная регургитация).
Конкретные признаки сильно различаются в зависимости от случая. Синдром Марфана наследуется как аутосомно-доминантный признак. Дефекты гена фибриллина-1 (N1) связаны с этой болезнью.
Лечение
Проблемы XYY синдрома устраняются при помощи различных видов терапии. Физиотерапия назначается для мальчиков с низким мышечным тонусом (гипотония), речевая при задержке речи.
Для мальчиков с лишней хромосомой может быть специальное школьное образование, или дополнительная помощь в некоторых классах.
Другие варианты лечения включают поведенческую терапию или препараты от гиперактивности дефицита внимания (СДВГ), поведенческих проблем.
Если присутствует расстройство аутистического спектра, применяется поведенческий анализ (АВА), рекомендована терапия. В некоторых случаях используется гормональная терапия. Устранение акне помогает самооценке пострадавшего человека.Лечение является симптоматическим и поддерживающим. Затронутые люди очень чувствительны к раннему вмешательству и лечению, проблемы могут полностью исчезнуть в течении нескольких лет.
Прогноз
Долгосрочная перспектива для мужчин с 47 хромосомами, как правило, хороша. Мальчики с этим синдром могут преуспеть как в школе, так и построении социальных отношений.
Лишняя хромосома у мальчиков не мешает построению успешной карьеры и созданию семьи. Терапия важна, так как позволяет затронутым людям полностью реализовать свои возможности.
По Рошель К. Ленрот, профессор, доктор медицинских наук, кафедры детской и подростковой психиатрии, Университет Нового Южного Уэльса.
Синдром Клайнфельтера: мужчина с женской хромосомой

Елена Шведкина об одном из самых распространенных генетических заболеваний — больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера — генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY. При этом в мужских половых железах — яичках — образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара — половая.
Женщины имеют пару половых хромосом ХХ, а мужчины — ХY.
Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х-хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY — наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х-хромосом, дополнительную Y-хромосому — 48,ХХYY. И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY/47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички.
Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E.
Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний: на каждые 500 новорождённых мальчиков приходится 1 ребёнок с данной патологией.
Кроме того, синдром Клайнфельтера — третья по распространенности эндокринная патология у мужчин (после сахарного диабета и патологии щитовидной железы) и наиболее частая причина врожденного нарушения репродуктивной функции у мужчин.
На сегодняшний день около половины случаев синдрома Клайнфельтера остаются нераспознанными. Часто такие пациенты обращаются за помощью по поводу бесплодия, эректильной дисфункции, гинекомастии, остеопороза, анемии и пр. без установленного ранее диагноза.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны.
Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще.
Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип.Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается.
Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде.
Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания.
В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит.
В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции.
У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью.
Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень.
Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции.
Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью.Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании.
Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
| Анализы | Результаты |
| Кариотип | 47,ХХY (80 % случаев) 48,ХХYY 48,ХХХY 49,ХХХХY 46,ХY/47,ХХY |
| Концентрация ЛГ, ФСГ | Повышена, особенно ФСГ |
| Концентрация общего тестостерона | Чаще снижена (в некоторых случаях нормальная за счет повышения секс-стероид-связывающего глобулина СССГ или на начальной стадии развития заболевания) |
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии — повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона.
Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза.
При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора — препарат-депо с замедленным высвобождением действующего вещества — инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
- денситометрию (всем пациентам, у которых на момент постановки диагноза были обнаружены остеопения или остеопороз).
Внедрение интрацитоплазматической инъекции сперматозоида в яйцеклетку (ИКСИ) и данные о возможности присутствия зародышевых клеток в яичках у пациентов с синдромом Клайнфельтера предопределили применение метода искусственного оплодотворения для данной категории пациентов, некоторые попытки были удачными.
Генетика — Онегомед ЭКО
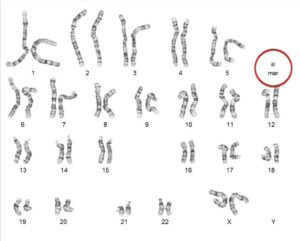
Ни одна современная клиника, оказывающая услуги ВРТ (вспомогательные репродуктивные технологии), не мыслит своей работы без использования различных генетических методов исследования, консультаций клинического генетика.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
ЗАЧЕМ?
Медико-генетическое консультирование проводится с целью:
- профилактики генетических аномалий у будущего потомства, основывающейся на данных о типе и варианте наследования патологического состояния, результатах преимплантационного генетического тестирования (ПГТ), пренатальной диагностики (НИПТ и инвазивная диагностика) и т.п.
- при подозрении на генетические аномалии — уточнения диагноза с использованием специальных генетических методов: генеалогическое обследование и составление родословной, биохимико-генетические методы, позволяющие выявить генетически обусловленные изменения обмена веществ, диагностика гетерозиготного носительства рецессивных аллелей, пренатальная диагностика (УЗИ, биохимический скрининг маркерных белков в сыворотке беременной, амниоцентез — забор околоплодной жидкости для кариотипирования плода)
- формулирования заключения и объяснение заинтересованным лицам в доступной форме смысла генетического риска
Каждая программа генетического обследования создается индивидуально, в зависимости от персонального и семейного анамнеза, медицинских задач, которые пациент ставит перед врачом-генетиком:
- цитогенетическое исследование (кариотипирование): выявление нарушения числа и структуры хромосом
- диагностика микроструктурных перестроек
- диагностика наследственных заболеваний
- выявление предрасположенности к различным заболеваниям, в том числе – раку молочной железы и яичников (для женщин) и прочих
- прогнозирование риска передачи заболеваний по наследству
- объяснение природы заболевания и типа наследования
КАК это происходит?
Медико-генетическое консультирование может быть как индивидуальным (взрослых и детей при подозрениях на генетическую патологию, беременных), так и семейным (когда консультация проводится паре, планирующей беременность).
В нашей клинике ведет прием опытнейший клинический врач-генетик высшей квалификационной категории Новожилова Тамара Семеновна
Биопсия трофэктодермы
ПРЕИМПЛАНТАЦИОННОЕ ГЕНЕТИЧЕСКОЕ ТЕСТИРОВАНИЕ эмбрионов (ПГТ)
ПГТ — это такое исследование, которое позволяет изучить хромосомный набор эмбрионов перед переносом их в полость матки, что позволяет перенести только тот эмбрион, который не имеет хромосомной патологии, могущей привести к выкидышу, неразвивающейся беременности или грубым порокам развития.
Хромосомные аномалии встречаются довольно часто. По статистике примерно половина из всех эмбрионов содержат одну или несколько хромосомных патологий. Это приводит к спо
нтанному прерыванию беременности на ранних сроках беременности, отсутствию наступления беременности и даже мертворождению.
Существуют несколько хромосомных патологий, которые могут привести к рождению живых детей с множественными нарушениями умственного и физического развития, например, при синдромах Патау, Эдвардса, Дауна и др.
У пациентов нашей клиники есть уникальная возможность проводить обследование полученных в циклах ЭКО эмбрионов на перечисленные выше хромосомные аномалии
Биопсия трофэктодермы
Для этого на 5 день культивирования производится биопсия и, несколько клеток будущих оболочек эмбриона берется на анализ.
На сегодняшний день наиболее передовыми являются aCGH (сравнительная полногеномная гибридизация на микрочипах) и NGS (секвенирование следующего поколения).
Исследуются все 23 пары хромосом!
Для кого показано ПГТ в первую очередь:
- Пациенткам после 35 лет.По статистике количество аномальных эмбрионов в 35лет с 50% к 39 годам достигает 75%, а к 40 годам – 85%!
- Парам с привычным невынашиванием беременности или замершими беременностями в анамнезе
- Мужчинам с тяжелыми нарушениями сперматогенеза
- Парам с множественными неудачными попытками ЭКО
- Пациентам с известными нарушениями кариотипа
При таком виде исследования сразу понятен пол плода, поскольку половые хромосомы также определяются
Статистически доказано, что у пациенток после 35 лет после ПГТ значительно увеличивается эффективность ЭКО и может достигать 65-70%!
НЕИНВАЗИВНЫЙ ПРЕНАТАЛЬНЫЙ ТЕСТ (НИПТ)
Такие тесты на сегодняшний день являются самыми безопасными и «продвинутыми» методами диагностики хромосомных аномалий плода.
В ЧЁМ СУТЬ?
В 9-11 недель забирается кровь у беременной, после чего происходит выделение хромосомной ДНК плода из крови матери, после чего становится возможным провести анализ структур
ных и количественных изменений хромосомного набора плода и предпринять необходимые меры. По сути метод является альтернативой инвазивным методам пренатальной диагностики и иногда может заменять их.
ПОКАЗАНИЯМИ для НИПТ являются:
- возраст будущей матери старше 35 летРабота в лаборатории
- подозрение на генетические нарушения у плода после проведения УЗИ и наличие биохимических маркеров, сигнализирующих о возможной хромосомной патологии
- наличие беременности, достигнутой при применении методов ВРТ
- наличие в анамнезе у беременной рождения мертвого ребенка (антенатальная гибель плода) или уже рожденного ребенка с пороками
- развития
- невынашивание беременности в анамнезе (выкидыши, неразвивающиеся беременности)
- наличие среди близких родственников ребенка хромосомным аномалиями и / или наследственными заболеваниями
- наличие кровно-родственного брака
- другие
В нашей клинике беременная может сдать кровь на НИПТ, предварительно (в 8-9 недель) высказав свое желание о необходимости проведения такого анализа.
КАРИОТИПИРОВАНИЕ*
КАРИОТИП — совокупность признаков (число, размеры, форма и т.д.
) полного набора хромосом, присущая клеткам данного биологического вида (видовой кариотип), данного организма (индивидуальный кариотип) или линии (клона) клеток.
Кариотипом иногда также называют и наглядное представление полного хромосомного набора (кариограммы), а сам метод исследования кариотипа называют кариотипированием.
ДЛЯ ЧЕГО?
Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у супругов при бесплодии или привычном невынашивании беременности. Выявление хромосомных перестроек в этом случае позволяет установить причину бесплодия и прогнозировать риск рождения в данной семье детей с хромосомной патологией.
Хромосомы
Современные методы кариотипирования обеспечивают:
- детальное обнаружение внутрихромосомных и межхромосомных перестроек,
- выявление нарушений порядка расположения фрагментов хромосом — делеции, дупликации, инверсии, транслокации
- диагностирование ряда хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме.
Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимы с жизнью. Это приводит к самопроизвольным выкидышам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности.
Примеры аномальных кариотипов и присущие им синдромы-заболевания:
Если кариотип не норма
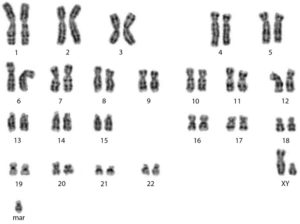
Патология в кариотипе влияет на процесс созревания половых клеток у мужчин и женщин, их качество и количество. Хромосомные перестройки в организме одного из супругов могут быть причиной бесплодия, повторяющихся выкидышей и рождения детей с пороками развития.
Частота обнаружения хромосомных перестроек составляет 1 случай на 100-200 пар. У таких людей повышенный риск гибели эмбриона, часто отмечаются нерезультативные циклы ЭКО. Что же делать, если пришёл результат анализа и оказалось, что кариотип не норма?
В ходе проведения анализа на кариотип чаще всего выявляются:
- синдром Шерешевского-Тернера – кариотип 45,Х;
- мозаичный вариант этого же синдрома – кариотип 45,Х/46,ХХ (только часть клеток несут в себе хромосомные аномалии);
- трисомия Х – кариотип 47,ХХХ;
- терминальная делеция длинного плеча Х-хромосомы – 46,ХХ,del (X)(q22);
- патологии аутосом (робертсоновская транслокация, лишняя маркерная хромосома).
Существует также множество других патологий: синдром Патау – кариотип 47,ХХ, 13 или 47,ХY, 13 (трисомия по 13 аутосоме), Дауна (трисомия по 21 хромосоме), Прадера-Вилли, Клайнфельтера и друге. Но они имеют явные клинические симптомы, поэтому случайное обнаружение у пар с бесплодием или проблемой привычного невынашивания перечисленных патологий маловероятно.
Что делать при выявлении хромосомных перестроек?
Если анализ на кариотип супругов показал патологию у одного из будущих родителей, требуется консультация генетика, чтобы понять, может ли человек быть генетическим родителем ребёнка. При некоторых хромосомных аномалиях зачатие невозможно, так как половые клетки у пациента попросту отсутствуют. В других ситуациях удаётся добиться беременности и рождения здоровых детей.
В любом случае беременеть лучше с использованием вспомогательных репродуктивных технологий. Они помогают снизить риск неудачных исходов беременности, рождения детей с хромосомными аномалиями или пороками развития внутренних органов. Для уменьшения вероятности подобных проблем используется ПГТ (преимплантационное генетическое тестирование).
Зачем делают ЭКО при хромосомных перестройках?
Во-первых, хромосомные перестройки часто становятся причиной бесплодия. Поэтому нередко супруги при всём желании не могут добиться беременности самостоятельно. Во-вторых, ЭКО и ПГТ позволяют снизить риск выкидыша и обеспечивают рождение здорового малыша.
Суть процедуры:
- Назначаются препараты для стимуляции суперовуляции.
- Женщине делают пункцию фолликулов и получают яйцеклетки.
- Они оплодотворяются сперматозоидами супруга методом ЭКО, ИКСИ или ПИКСИ (в зависимости от качества спермы).
- Эмбрионы культивируют в лаборатории.
- На пятый день у эмбриона берутся клетки для проведения преимплантационного генетического тестирования.
- Эмбрионы замораживаются на хранение до переноса.
- После получения результатов преимплантационного генетического тестирования эмбрионы, имеющие хромосомные перестройки, отбраковываются.
- В следующем цикле женщине делают перенос «здорового» эмбриона, в котором по данным ПГТ отсутствуют хромосомные перестройки.
Такой подход позволяет в большинстве случаев добиться наступления беременности, успешно выносить и родить здорового ребёнка.
Использование донорского биоматериала
В некоторых случаях ПГТ не позволяет решить проблему. Иногда половых клеток нет вообще. В иных случаях все они несут дефектный генетический материал, поэтому добиться получения жизнеспособных эмбрионов невозможно. В той ситуации используют донорскую сперму или ооциты.
Если проблемы с хромосомным набором обнаружены у мужчины, для оплодотворения используют искусственную инсеминацию. Сперму донора вводят при помощи тонкого катетера – гибкой трубки, помещенной в матку. Это делают 1 или 2 раза в течение цикла, в наиболее фертильные дни. Если беременности нет, процедуру повторяют в следующем цикле.
При наличии хромосомных перестроек, несовместимых с беременностью, у женщины, показано ЭКО с донорскими ооцитами. Половые клетки оплодотворяются спермой супруга. Затем они переносятся в матку. С высокой вероятностью в результате этой процедуры наступит беременность, которая будет нормально развиваться и закончится родами.
Поэтому не стоит отчаиваться, если кариотип – не норма. Современная медицина имеет в своём арсенале способы помочь паре зачать и родить здорового ребёнка.
Автор текста: Врач-эмбриолог, генетик, к.м.н. Митюшина Н.Г.
Стоимость анализа кариотипа
Все услуги
| Анализ кариотипа 1 пациента (кровь с гепарином, срок дни — до 16-20 раб. дн.) | 4 730 руб. |
| Анализ кариотипа (с фотографией хромосом) 1 пациента (кровь с гепарином, срок дни — до 16-20 раб. дн.) | 5 250 руб. |
| Кариотипирование с выявлением аберраций (кровь с гепарином, срок дни — до 16-20 раб. дн.) | 6 090 руб. |
| Кариотипирование с выявлением аберраций (с фотографией) (кровь с гепарином, до 16-20 раб. дней) | 6 620 руб. |